








Our Company
THE LEADING RNAi THERAPEUTICS COMPANY
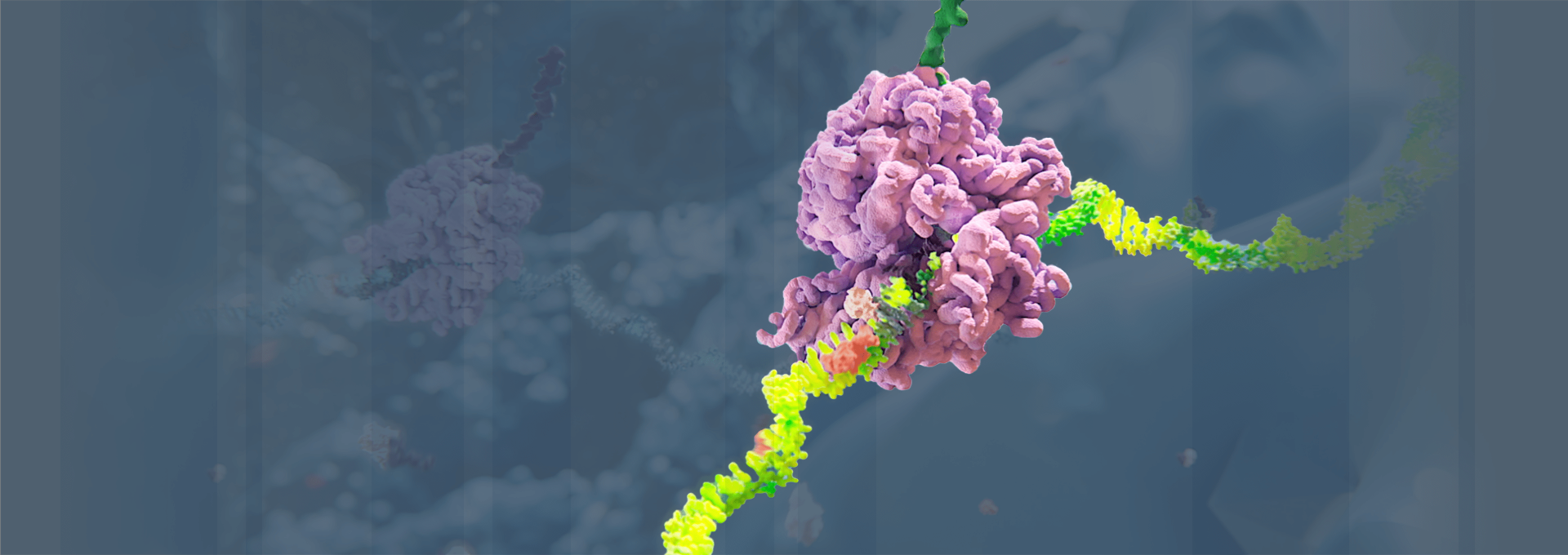
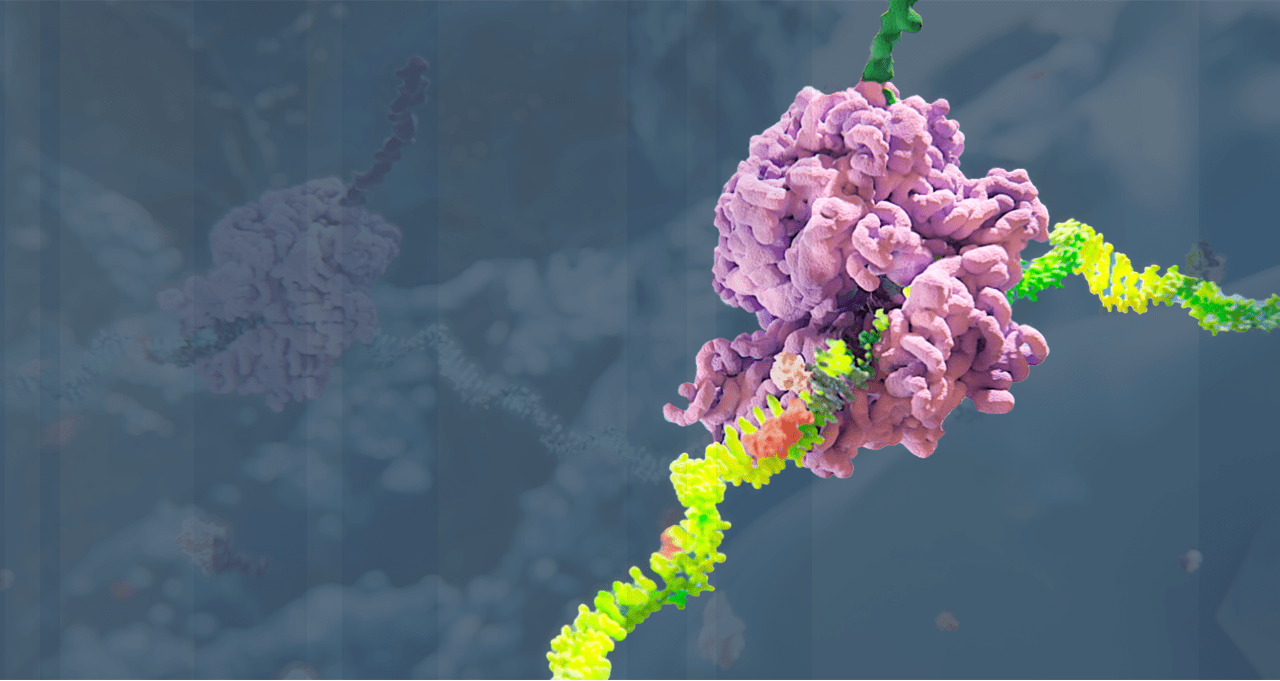
Alnylam has led the translation of RNA interference (RNAi) from Nobel Prize-winning discovery into an entirely new class of medicines, which we believe has the potential to help people all over the world to live longer, healthier, and fuller lives. Through pioneering science, unwavering commitment to patients, and our approved therapies and deep pipeline of investigational RNAi therapeutics, we have cemented our reputation for innovation and leadership in RNAi.
OUR SCIENCE IS CHANGING THE WAY
MEDICINE TREATS DISEASE™
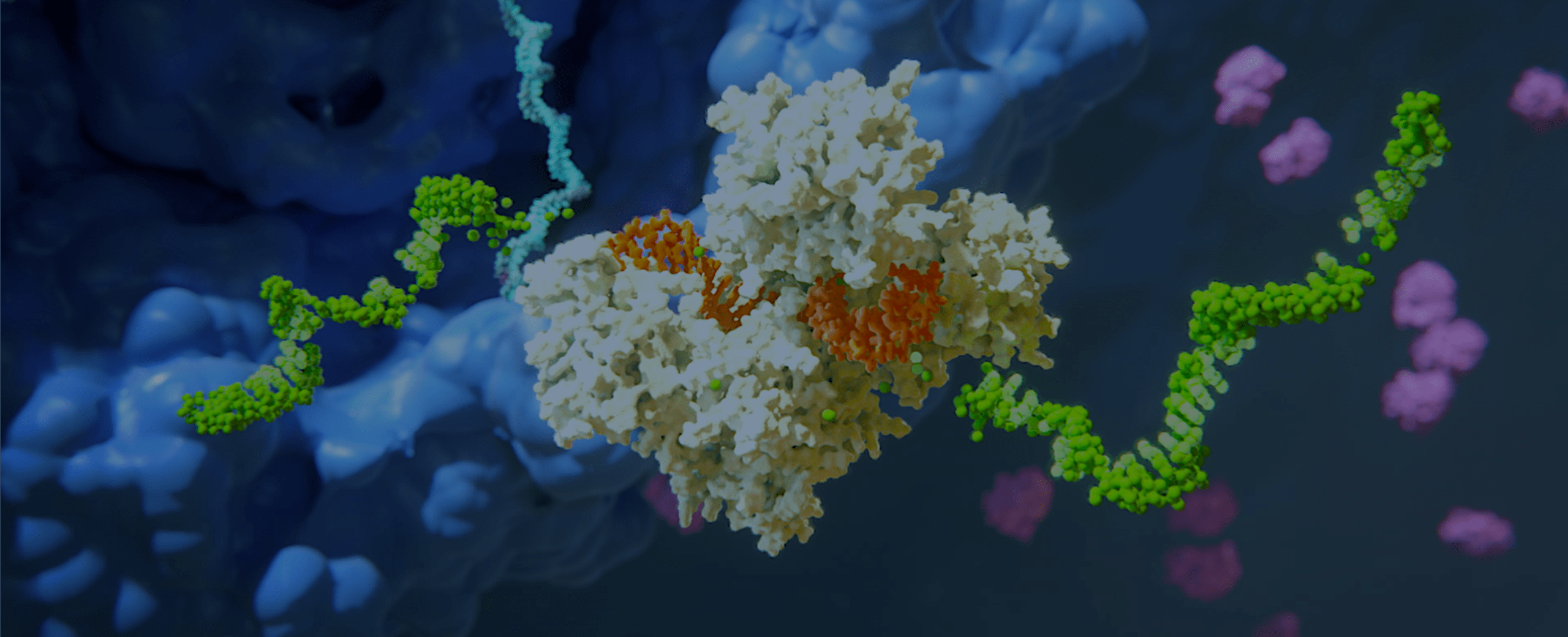
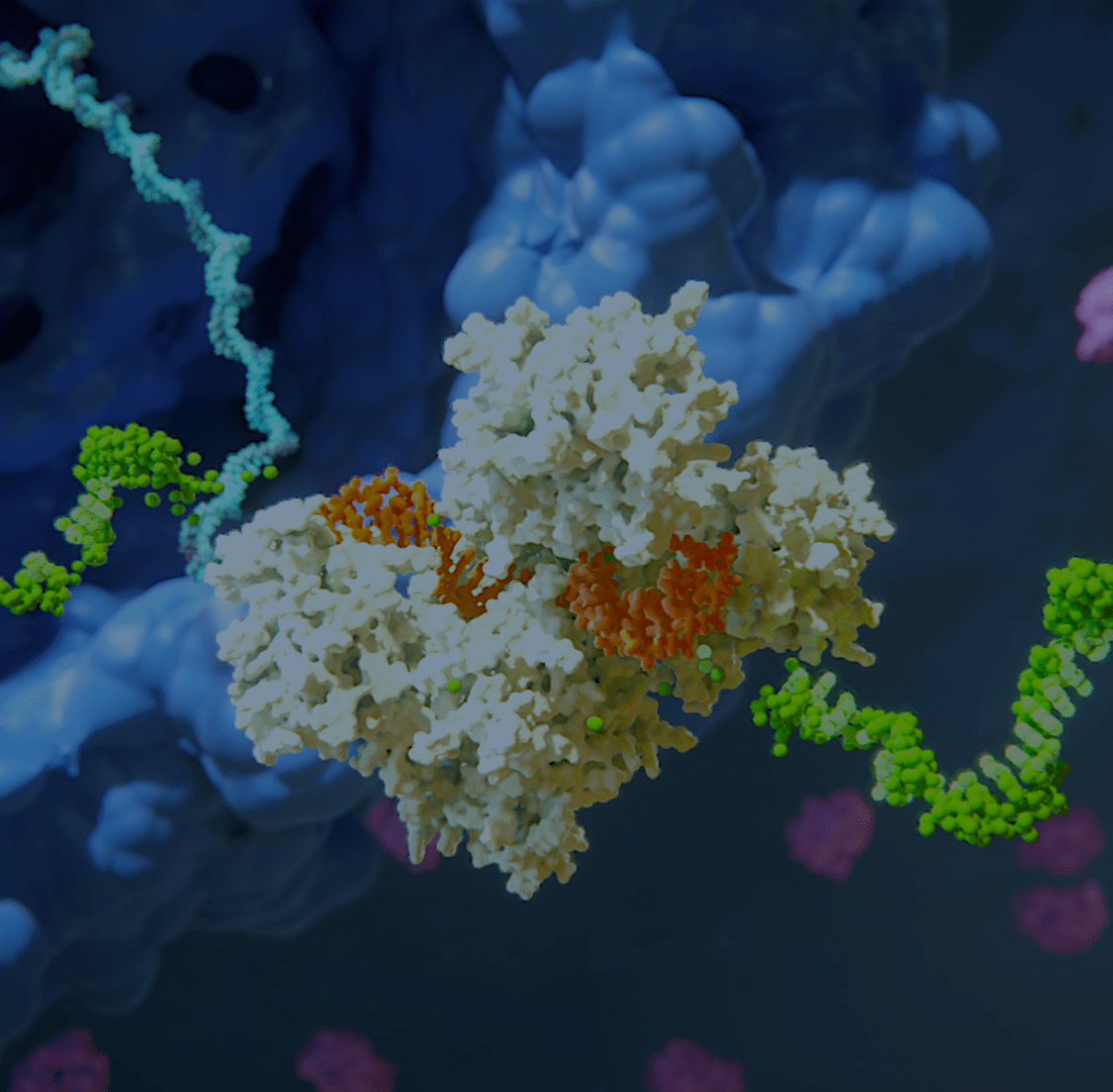
Our Science
OUR PIPELINE
Our robust pipeline of investigational RNAi therapeutics is focused on diseases with high unmet medical need which fall into four main therapeutic areas: genetic medicines, cardio-metabolic diseases, infectious diseases, and central nervous system (CNS) and ocular diseases. We have more than a dozen RNAi therapeutics in clinical development, including multiple in late-stage development.
Genetic Medicines
Cardio-Metabolic Diseases
Infectious Diseases
Central Nervous System & Ocular Diseases
PATIENTS COME FIRST
Whether we're working to develop the first treatment for a disease or a better one, patients are the center of all we do. They are the reason we come to work every day, and their stories inspire us to continue to innovate by pushing the science of RNAi therapeutics forward.

My body started to warn me that something was wrong. That I had a problem… My father passed away 40 years ago; after I received my diagnosis, we believe what he really had was hATTR amyloidosis like me.
-Chen, living with hATTR amyloidosis
ALNYLAM NEWS





Featured Articles

Corporate Responsibility
We accept bold challenges to improve the health of humanity.
We believe that our actions as a company and employees can be a force for good in the world. Consistent with our core values, we are relentless advocates for science, patients, employees, communities, and our planet.
Our Company
Diversity, Equity & Inclusion
We are proud of the award-winning, open, dynamic, and diverse culture we have built, where employees feel included, supported, and heard. We are committed to building a work environment with a shared mission and values, where all employees feel they belong and are enabled to achieve their full potential.


This is your moment to join us
We're 2,000+ smart, passionate, "change the world" kind of people who believe deeply in the power and potential of our technology and what we're building as a company. If you are looking for a mission and colleagues that inspire and a company that recognizes and rewards both group and individual contributions, perhaps this is your moment to join us.
JOIN US ›